
材料シミュレーション技術が集約された原子スケール材料シミュレータ「matelier」を、技術者・研究者の方々に使いやすい形でご提供いたします。現在「matelier」で、第一原理計算による電子状態計算を行う、第一原理バンド計算ソフト「PHASE/0」をご利用いただけます。サポートサービスに力を入れておりますので、初めて材料シミュレーションに取り組むお客様にも安心してご利用いただいています。
![]()
![]()
PHASE/0は、平面波基底を採用した密度汎関数法による第一原理擬ポテンシャルバンド計算ソフトウェアです。

第一原理計算では量子力学に則って物質の電子状態を求めますので、精密な解析が可能です。計算に際して、実験結果に基づくパラメータは必要ありません。実験結果の解釈や、新規材料の物性値予測にもご活用いただけます。
基本機能: エネルギー / スピン分極 / 状態密度 / 原子に作用する力 / 格子に作用する応力
実空間量: 安定な原子配置 / 電荷密度分布
波数空間量: バンド構造図 / フェルミ面(等エネルギー面)
電子状態解析: 局所状態密度 / 射影状態密度
分子動力学: NVTおよびNVE
遷移状態探索: Nudged Elastic Band法
その他: 仕事関数 / 誘電関数(格子系、電子系) / 圧電定数 / STM像 など
![]()
![]()
第一原理バンド計算PHASE/0は擬ポテンシャル法を採用しており、内殻電子をあらわに取り扱うことなく価電子の状態のみを精密に求めます。擬ポテンシャルは、あらゆる環境下で実際の原子の如く振る舞うように、第一原理計算に基づいて作成されています。
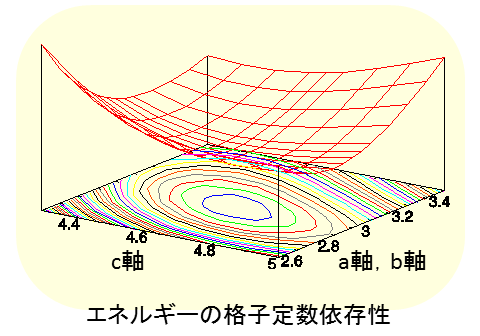
ここでは、擬ポテンシャルが異なる環境下でも原子のように振る舞うことを確認するために、酸化チタンの計算を行いました。まず、ルチル型のTiO2を取り扱います。エネルギーの格子定数依存性を計算することにより、エネルギーが最も低くなる、すなわち、安定な格子定数を求めました。計算の結果、a = b = 4.66 (Å), c = 2.98 (Å)となり、実験値と良く一致することが確認できました。
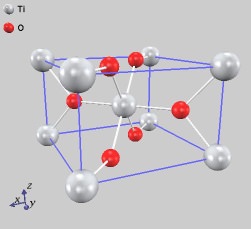
ルチル型
同様の計算を、結晶構造の異なるTiO2(アナターゼ型とブルッカイト型)さらには還元型酸化チタンTi2O3に対しても行いました。計算結果を実験値とともに表にまとめます。どの構造に対しても計算結果は実験値と良く一致しており、計算では「擬ポテンシャル」として扱っている各原子が実在の原子のように振る舞っていることが確認できます。
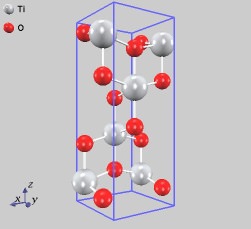
アナターゼ型
ブルッカイト型
還元型Ti2O3
| 格子定数 (Å) | |||||||
|---|---|---|---|---|---|---|---|
| a軸 | b軸 | c軸 | |||||
| 計算値 | 実験値 | 計算値 | 実験値 | 計算値 | 実験値 | ||
| TiO2 | ルチル | 4.66 | 4.59 | ← | 2.98 | 2.96 | |
| アナターゼ | 3.82 | 3.78 | ← | 9.74 | 9.51 | ||
| ブルッカイト | 9.32 | 9.17 | 5.52 | 5.45 | 5.17 | 5.14 | |
| Ti2O3 | 5.13 | 5.15 | ← | 14.06 | 13.54 | ||
PHASE/0の計算では電子状態を精密に求めていますので、状態密度やバンド構造を得ることができます。例として、MgB2の状態密度とバンド構造を図に示します。バンド構造図作成の際にはGUIを利用して、ブリルアンゾーン内の対称線を指定しました。
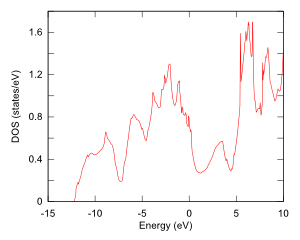
状態密度

バンド構造
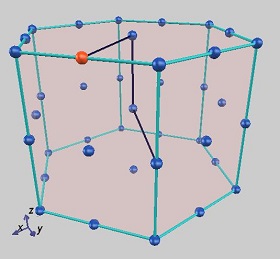
GUIを用いた対称線の指定
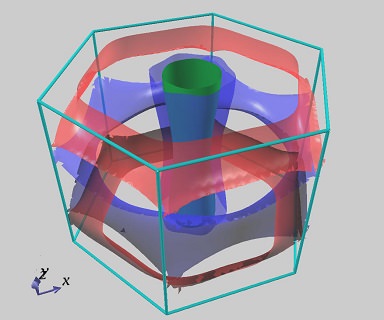
また、フェルミ面(等エネルギー面)を描画することもできます。
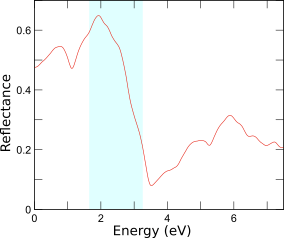 可視域
可視域
黄銅鉱の反射率
黄銅鉱(CuFeS2; chalcopyrite)は反強磁性半導体ですが、見た目が「金ピカ」であることが知られています。その性質を調べるために電子状態から誘電率を求め、反射率のエネルギー依存性を計算しました。可視光線の低エネルギー成分(赤)を比較的良く反射しますが、エネルギーが高くなると急激に反射率が低下することがわかります。この振る舞いは本物の金と似ており、黄銅鉱が「金ピカ」に見えることが理解できます。
上の計算は電子系誘電率(バンド間遷移起因)ですが、誘電率にはその他に格子系(フォノン)からの寄与も存在します。可視域では、その電磁波の速い振動に原子が追従できないため格子誘電率は無視できますが、赤外などエネルギーが低い(低振動数)領域ではフォノンが誘電率に影響を及ぼします。電子材料に対しては、格子系まで含めた誘電率の評価が大切です。
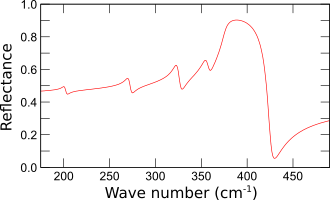
黄鉄鉱の赤外反射率
格子誘電率計算の例として、同じく見た目が金と似ていることで有名な黄鉄鉱(FeS2; pyrite)の赤外反射率の波数依存性を示します。フォノンの振動数で特徴的な振る舞いをしています。また、LO-TO分裂を考慮した解析であることがわかります。
さらに、赤外分光の相補的な手法であるラマン分光につきましては、こちらをご覧ください。
密度汎関数法では、電荷密度が非常に重要な物理量であり、その計算結果を可視化することができます。例としてグラフェン(graphene)の電荷密度分布を下図左に示します。炭素間の化学結合に沿って電子が分布していることが分かります。さらに、特定のエネルギー領域の電荷密度だけを抜き出して可視化することも可能です。その例として、グラフェンのフェルミエネルギー付近の電荷密度のみを可視化しました(下図右)。グラフェンの伝導を担っているのはπ電子であることを示唆しています。
グラフェンの電子状態解析につきましては、こちらも併せてご覧ください。

全電荷密度

フェルミエネルギー付近の電荷密度
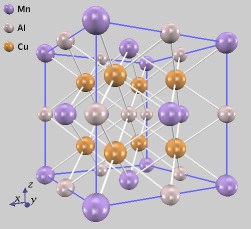
ホイスラー合金の顕著な性質の一つに、非磁性元素のみを用いて強磁性体を得られることが挙げられます。その例として、Cu2MnAlの電子状態を計算しました。
常磁性および強磁性状態について、安定構造のエネルギーを求めました。その結果、常磁性状態よりも強磁性状態の方がエネルギーが低く、安定であることがわかります。さらにそのときの格子定数は、実験値と非常に良く一致しています。
| 格子定数 (Å) | エネルギー* (eV/atom) | ||
|---|---|---|---|
| 計算値 | 常磁性 | 5.85 | |
| 強磁性 | 5.96 | -0.27 | |
| 実験値 | 5.97 | ||
PHASE/0の計算では相関交換エネルギーに対する近似として、GGA (Generalized Gradient Approximation)を用いるのが標準的です。この手法により、比較的少ない計算負荷で数多くの物性値を精度良く求めることに成功してきました。この成功があったからこそ、第一原理バンド計算がここまで普及したといっても過言ではありません。しかしながら、GGAで全ての物理量を精度良く求めることができるわけでもありません。不得手な物理量の代表が「バンドギャップ」です。この欠点を補うために数多くの研究がなされ、最近ではハイブリッド汎関数近似が実用的に使われるようになりました。その効果を、シリコンおよびスズの単結晶(ダイヤモンド構造)の計算結果を使って紹介します。
GGAの計算では、シリコンのバンドギャップは実験値の6割程度に過小評価されており、スズに至ってはバンドギャップが潰れて金属的な電子状態が得られました。一方、ハイブリッド汎関数(HSE06)を用いた計算では、実験値を再現するバンドギャップが得られます。それだけでなく、体積弾性率やフォノン振動数の実験値との一致も改善されることが確認できました。
ハイブリッド汎関数の応用事例として、光触媒材料のバンドギャップ解析も併せてご覧ください。
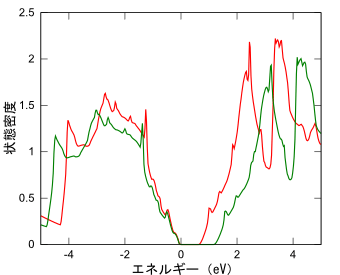
シリコン単結晶の状態密度
赤:GGA、緑:ハイブリッド(HSE06)
| シリコン | スズ | |||||
|---|---|---|---|---|---|---|
| GGA | HSE06 | 実験値 | GGA | HSE06 | 実験値 | |
| バンドギャップ (eV) | 0.64 | 1.18 | 1.11 | 0 | 0.02 | 約0.1 |
| 体積弾性率 (GPa) | 87.6 | 96.5 | 98.9 | 37.2 | 42.4 | 42.6 |
| フォノン振動数 (cm-1) | 502 | 526 | 520 | 179 | 196 | 199 |
ダイヤモンドはバンドギャップや絶縁破壊電圧が大きく、熱伝導率が高い、耐熱性に優れるなど、パワーデバイス用の材料として優れた特性を有しています。また、NVセンターを量子ビットに見立てたデバイス開発も含めて、次世代デバイス材料として幅広く研究されています。さらにもう一つ、ダイヤモンドの顕著な特徴に「負性電子親和力」が挙げられます。これは、真空準位が伝導帯の底よりも低いことを意味しており、伝導体に励起された電子は自ら真空中に出ることを好みますので、電子線源として利用できます。PHASE/0の仕事関数計算機能を利用してこれを解析しました。
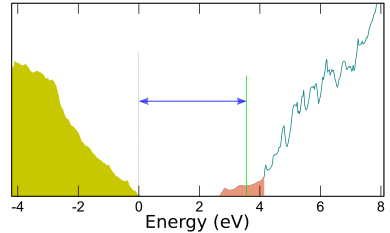 仕事関数
価電子帯
真空準位
表面
仕事関数
価電子帯
真空準位
表面
電子状態
水素終端ダイヤモンド(001)の状態密度と真空準位
(001) 2x1水素終端表面スラブモデルを作成し、構造緩和を含む電子状態計算を行いました。仕事関数計算機能を利用すると真空準位が求まりますので、その値を状態密度とともに示します。真空準位より低いエネルギーに伝導体の電子状態がありますが、部分電荷密度の解析から、これらは表面に局在していることが分かりました。すなわち価電子帯のバルク状態は全て真空準位より高いエネルギーにあり、電子親和力が負になることが確かめられました。
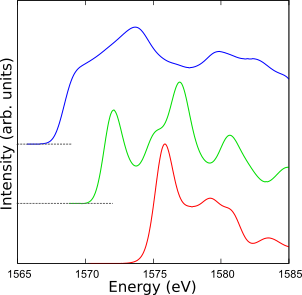 Al
Al2O3
AlN
Al
Al2O3
AlN
XANESスペクトル(Al K端)
内殻電子は化学結合にほとんど影響を及ぼしませんが、そのエネルギー準位は周囲の影響を受けています。 そしてX線などの高エネルギー電磁波を物質に照射すると内殻電子が励起されることがあり、 散乱されたX線を解析すると、放出源原子の電子状態を知ることができます。 ところで、擬ポテンシャル法では内殻電子を「ポテンシャル」に置き換えていますので、一見、このような解析には適さないように思われますが、 CIAOで作成する「内殻正孔を有する擬ポテンシャル」を用いることにより、取り扱い可能です。
X線吸収端近傍構造(XANES)の計算例を示します。アルミニウムのK端について、単体金属、窒化物と酸化物のスペクトルを比較しました。 物質の特徴がスペクトルに現れており、吸収端エネルギー位置が10 eV程度一様にシフトしていることを除き、実験を再現しました。
他に、X線光電子分光(XPS)や、電子エネルギー損失分光(EELS)の解析にも対応しています。
近年目覚しい計算機の性能向上に伴い、第一原理分子動力学計算(MD)にも手が届くようになりました。古典MDで正しい結果を得るためには、質の良い経験的な原子間ポテンシャルが必要ですが、常に用意できるものではありません。一方、第一原理MDでは電子状態から原子間に作用する力を計算するので、経験的な原子間ポテンシャルは不要です。
ここでは例として、固体酸化物型燃料電池(solid oxide fuel cell; SOFC)で電解質(O2-透過膜)として用いられているイットリア安定化ジルコニア(YSZ)の第一原理MDを行いました(温度:1,500K)。この物質には酸素空孔があり、そこに隣の酸素(O2-)が入ることによりO2-が移動します。その一方でイットリウム(Y)やジルコニウム(Zr)は格子点の周囲で振動するだけであり、固体としての骨格を保ちます。このような性質により、O2-を選択的に透過する膜として利用できるのです。
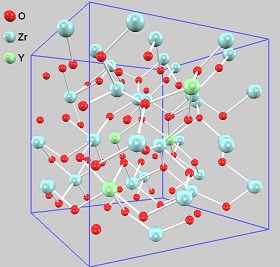
原子配置のスナップショット
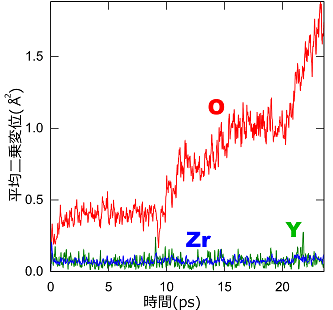
平均二乗変位
PCクラスターでは、ノード間がギガビットイーサネットなどで接続され、全てのノードはsshで相互に通信できることが必要です(数ノード程度であればギガビットイーサネットで並列性能発揮します)。
複数の計算ノードを利用した並列計算には対応していません。
メモリ搭載量は、2GB/コアを目安にしてください(例:4コア1CPUの計算機に8GB搭載)。大規模一点計算(構造最適化なし)や中規模系でのハイブリッド汎関数利用など、多くのメモリを必要とする計算でも4GB/コア程度をお勧めします。
ハイパースレッディングによる性能向上は大きくありません。
Linux機へのリモートジョブ投入などを行う場合には、Linux機がssh接続を受け付けることが必要です。VNC、リモートデスクトップ(Windows)、Xサーバーのリモート利用(Xクライアントと別計算機で動作するXサーバー)は動作保証対象外です。
「第一原理バンド計算ソフトウェアPHASE/0」
下記より製品資料をダウンロードいただけます。
![]()
![]()
![]()
![]()