
古典分子動力学は、ニュートンの運動方程式(古典力学)に従って原子の運動を追跡する計算手法です。原子間の相互作用には、経験的なポテンシャルを利用します。そして古典分子動力学の正確性は、このポテンシャルの精度にかかっています。原子間の相互作用は、材料の多様性を生み出す源ですので、単純な関数ではありません。例えば二原子間の距離のみに依存するポテンシャルは簡素で計算しやすいのですが、実用材料を記述するには表現力が不足しています。距離だけでなく、三つの原子の成す角度(結合角度)の影響を取り入れるなど、種々の工夫を取り入れたポテンシャルが提案され、それらを利用した多数の成功事例が報告されています。
以下では、解析対象に適したポテンシャルを使用した古典分子動力学シミュレーション解析事例をご紹介します。計算実行にはLAMMPSを用いました。
炭素原子は最外殻軌道2pに2個の電子を持ちますが、2価ではなく、4価であるように振る舞うことが知られています。そしてこの振る舞いの理解には、電子状態に基づく混成軌道の考え方が役立ちます。一方、古典分子動力学では電子状態を解くことなしに、sp2やsp3混成軌道を表現することが要求されます。これに応えるために、ポテンシャルの結合角度依存性に対して「結合次数(ボンドオーダー)」を導入することが提案されました。この考え方に基づくTersoffポテンシャルを用いて、アモルファスカーボン作成シミュレーションを行いました。


左:50 GPa、右:300 GPa
赤はsp2炭素、灰はsp3炭素
炭素材料に限らず分子動力学計算を使ってアモルファス構造を作成する際には、物質を高温で溶融した後に、冷却して固化させます。急冷すれば乱雑度が高く、ゆっくり冷却すれば結晶性が高くなるなど、冷却条件により異なる特徴を持つアモルファス構造が生成されます。ここでは降温時の圧力条件を変えた複数のアモルファスカーボンを作成しました。
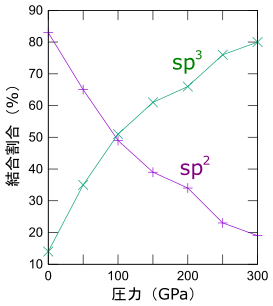
sp2, sp3の各割合
アモルファスカーボンはダイヤモンドライクカーボン(DLC)とも呼ばれ、製品の高品質化・高機能化に寄与する表面改質技術として、様々な技術分野で利用が広がっています。その物性を特徴づける代表的な指標が、sp2結合とsp3結合の比率です。そこで作成したアモルファス構造について、sp2結合とsp3結合それぞれの割合を求めました。それらの冷却時圧力依存性を左に示します。高圧下で冷却するほど、sp3結合の割合が高い、よりダイヤモンド的なDLCを作成することができました。

Li1+x+yAlxTi2-xSiyP3-yO12原子配置の
スナップショット(x = 1/3, y = 1/6)
リチウムイオン電池の必須構成要素は、電極(正極/負極)と電解質です。現在は電解質に有機溶媒を用いることが主流ですが、これを固体に替えて電池全体を固体にすること(全固体電池)により、安全性向上・大容量化・高速充放電など、様々なメリットを享受できることが期待されています。ここでは、リチウムイオン電池用固体電解質の例としてNASICON型Li1+x+yAlxTi2-xSiyP3-yO12について、リチウムイオン伝導(拡散)シミュレーションを行いました。一見、複雑な組成ですが、LiTi2(PO4)3のTiの一部(x)をAlに、Pの一部(y)をSiにそれぞれ置換し、価数が整うようにLiを追加したものです。以下ではx=1/3, y=1/6としました。
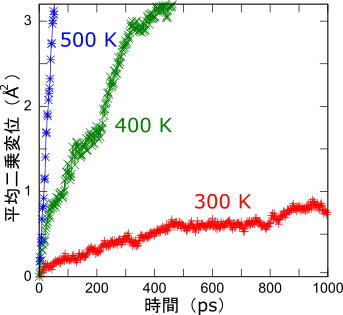
平均二乗変位
原子間の相互作用には、Pedoneポテンシャルを利用しました。このポテンシャルはLi0.6+, O1.2-, Si2.4+などのように各元素のイオン化度が設定されており、イオン間のクーロン相互作用を考慮します。
温度を300K, 400K, 500Kに設定してNPTシミュレーションを行い、平均二乗変位を求めました。結果を左に示します。直線で近似した傾きから求めたリチウムイオン伝導度は10-3 S/cm程度(300K)であり、実験結果とおおむね一致することが確認できました。
また本物質は1,000Kの高温に晒されてもLi以外の原子は移動せず、結晶の骨格は安定でした。この振る舞いは、後述する有機電解液と対照的です。
先のPedoneポテンシャルは、原子の電荷を考慮してはいるものの、その値はシミュレーション中変化しない固定値でした。リチウムイオン拡散現象では、明瞭な化学結合の組み換えが生じないため、電荷値が固定されているポテンシャルに特段の不都合は感じられませんでしたが、原子間結合、特に共有結合の組み換えが生じる過程を表現する際には、電荷値が固定されていることは、おそらく適切でないでしょう。そして古典分子動力学では電荷を可変にする手法(電荷平衡法)がすでに確立されています。
パワーデバイス用の半導体として、ワイドギャップ半導体SiCの実用化が進められています。パワーデバイスの材料としてSiCの素性の良さに疑いの余地はなく、高耐圧と低抵抗を両立できるMOSFETは、電源やインバータなどに利用されています。ここではSiC MOSFET作製の主要プロセスの一つであるゲート酸化膜形成を、電荷平衡法を利用してシミュレーション実行しました。
1,050Kに加熱したSiC(1120)基板に、高速の酸素分子をぶつけます。大きな運動エネルギーを持つ酸素分子は解離して原子となり、高温のSiC表面と反応します。原子間結合の組み換えを繰り返しながら、また、酸化炭素が脱離する過程などを経て、SiO2酸化膜が成長する様子を動画で示します。
本計算では、公開されているポテンシャルを使用しました。このポテンシャル作成の際には、第一原理計算PHASE/0の計算結果が利用されたそうです。
電荷平衡法を用いると計算負荷が高くなりますので、常に使えば良いという類のものではありません。適切なポテンシャルを選択することは、古典分子動力学シミュレーションを成功に導く重要なポイントです。
原子間結合の生成と解離を取り扱う反応力場として開発された原子間ポテンシャルがReaxFFです。ReaxFFは電荷移動はもとより、結合の二面角依存性など各種相互作用が取り入れており、化学反応など複雑な現象を再現できるように設計されています。ここではReaxFFを用いて、リチウムイオン電解液(溶媒)のシミュレーションを行いました。
上述の通り、リチウムイオン電池の必須構成要素は、電極(正極/負極)と電解質です。全固体電池の研究開発が進められていますが、実用化されている電池では液体の電解質、すなわち電解液が主流です。リチウムイオン電池は充放電過程で少なからず発熱しますので、電解液はある程度の高温まで安定に存在することが大切です。
溶媒シミュレーション
典型的な電解液は、電解質としてLiPF6等のリチウム塩を溶媒に溶解させたものです。溶媒には、エチレンカーボネート(EC)が良く使われます。ECは単なる溶媒ではなく、カーボン負極との間にSolid Electrolyte Interface (SEI)と呼ばれる保護膜を形成する作用もある、重要な材料です。一方純粋に溶媒としてみると、ECは粘度が高いことが欠点であり、実用的にはジメチルカーボネート(DMC)などの低粘度溶媒を混合して利用します。そこで、ECとDMCの1:1混合物の熱安定性を調べました。
まず室温(300K)でのシミュレーション結果を示します。ECとDMCいずれも変化することなく、安定に存在し続けます。
生成分子数(個)
| 300K | 1,000K | |
|---|---|---|
| CO2 | 0 | 29 |
| C2H4 | 0 | 10 |
| CH2O | 0 | 5 |
| CH4O | 0 | 2 |
次に高温(1,000K)でのシミュレーションを行いました。溶媒の一部が分解して、複数種の化合物が生成されました。250ps後の生成分子数を左表に示します。生成物に、二酸化炭素CO2やエチレンC2H2など(室温で)気体が含まれることに注目してください。液体から気体になると、体積が著しく増加します。現実世界では、リチウムイオン電池は密閉容器に納められていますので、この容器が膨らみます。シミュレーション条件は極端な高温ですが、シミュレーション総時間は1ナノ秒(10-9秒)に過ぎません。リチウムイオン電池は数時間~日の単位で利用するデバイスです。実使用時に相当する温和な温度条件下では上記計算結果と比較して分解頻度が大幅に減少しますが、動作が長時間に及ぶことを併せて考慮すると、分解の可能性を認識することは重要です。

断熱シミュレーションの温度変化
さらに、断熱シミュレーションも試みました。極短時間(0.5ピコ秒 = 5×10-13秒)1,000Kでシミュレーション実行した後、温度制御を止めてNVEシミュレーションに移行しました。その結果、溶媒が分解する際の発熱で温度が上昇、それがさらなる分解を促し、発火に相当する急激な温度上昇が認められました。電解液を用いた電池では大きな機械的な力が作用した場合などに正極と負極が短絡する恐れがあり、それが原因で大電流が生じ、瞬間的にでも高温が発生すれば、このシミュレーション結果が現実になりかねません。これを回避するために、電極はセパレータと呼ばれる膜で隔てられています。一方、機械的強度が向上する全固体電池では短絡の可能性がほとんどなくなり、セパレータを利用せずに安全性を確保できることが期待されています。なお、現実のリチウムイオン電池は断熱状態ではありませんが、再度、シミュレーション時間が極めて短いこと(ここでは250ピコ秒 = 2.5×10-10秒)を思い出してください。現実的な熱伝導条件では、これほどの短時間に放出する熱量は少なく、それを無視する断熱条件は妥当な近似です。
古典分子動力学よる受託解析を承っております。
アモルファス構造作成など第一原理計算の前処理として、もしくは、結晶成長プロセスなどの大規模長時間の分子動力学シミュレーションに、ポテンシャルの選定からご相談を承ります。ぜひ一度お問い合せください。
![]()
![]()
![]()